

CRISPR-Cas System: The Current and Emerging Translational Landscape
 ,
,
Abstract
1. Introduction
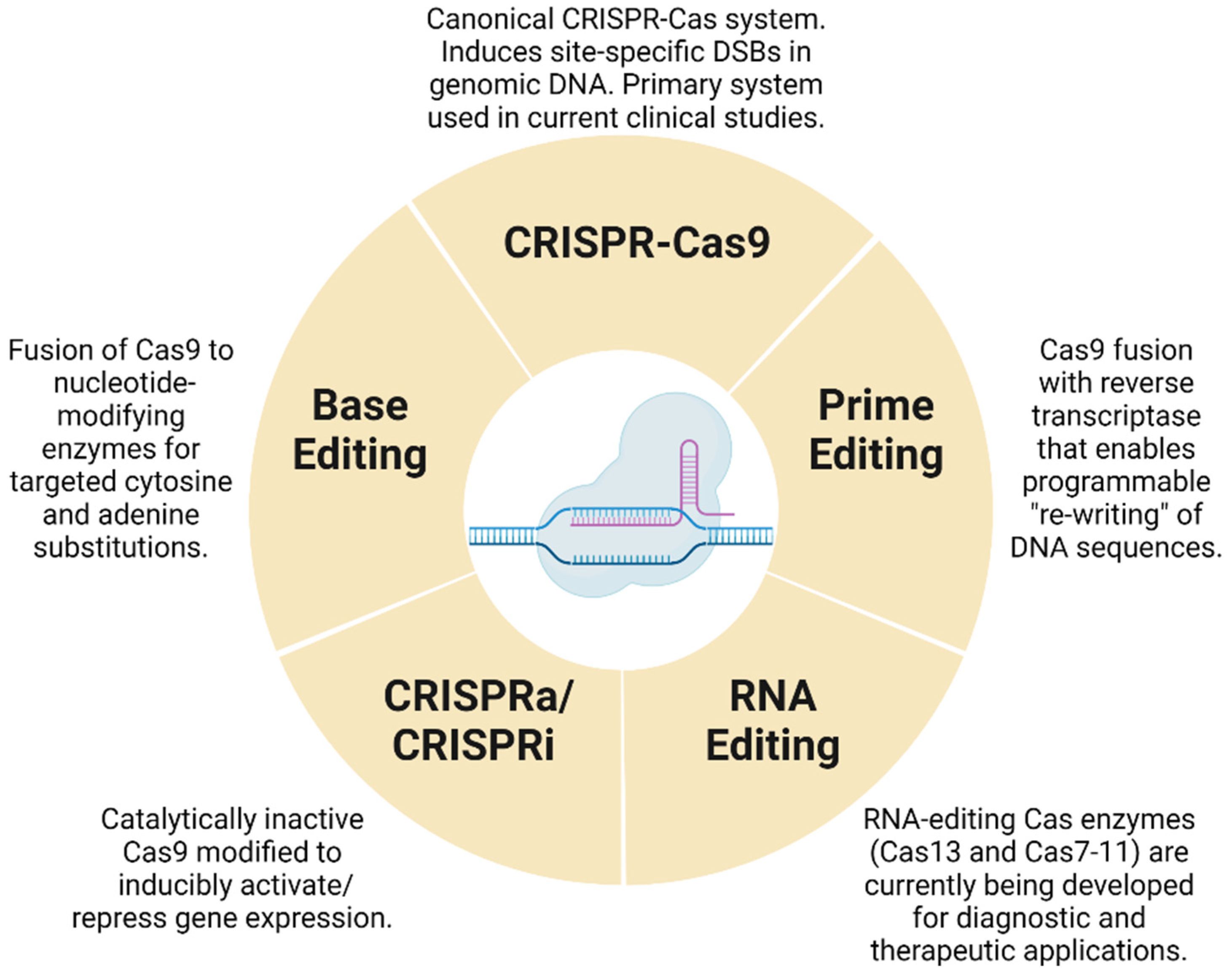
2. Current Use of CRISPR-Cas Systems in Clinical Trials
2.1. CRISPR-Cas Technologies in the Treatment of Cancers
2.1.1. Cellular Immunotherapies
2.1.2. Role of CRISPR-Cas9 Tools to Mediate Immune Check Point Inhibition in CAR-T Therapy

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Conditions Targeted | Targets Knocked out via CRISPR | Targets Knocked in (via Lentivirus or CRISPR) | Sponsor | Clinical Trial ID |
|---|---|---|---|---|
| Advanced hepatocellular carcinoma | PD1 | Central South University | NCT04417764 [43] | |
| Advanced esophageal squamous cell carcinoma | PD1 | Hangzhou Cancer Hospital | NCT03081715 [44] | |
| Metastatic gastrointestinal cancers | CISH | Intima Bioscience, Inc. | NCT04426669 [60] | |
| Metastatic non-small cell lung cancer | NCT05566223 [59] | |||
| Metastatic non-small cell lung cancer | PD1 | Sichuan University | NCT02793856 [45] | |
| EBV+ malignancies | PD1 | Nanjing University | NCT03044743 [46] | |
| CD5+ relapsed/refractory T cell malignancies | CD5 | CD5-CAR (via lentivirus) | Huazhong University | NCT04767308 [61] |
| Acute lymphocytic leukemia | HPK-1 | CD19-CAR (via lentivirus) | Xijing Hospital | NCT04037566 [62] |
| Multiple solid tumors | PD1 and TRAC | Mesothelin-CAR (via lentivirus) | Chinese PLA General Hospital | NCT03545815 [47] |
| Mesothelin-positive multiple solid tumors | NCT03747965 [48] | |||
| Advanced EGFR-positive solid tumors | TGF-β receptor Ⅱ | EGFR-CAR (via lentivirus) | NCT04976218 [63] | |
| Multiple myeloma | PD1, TRAC and TRBC | NY-ESO-1-TCR (via lentivirus) | University of Pennsylvania | NCT03399448 [49] |
| Acute myeloid leukemia | TRBC and TRAC | Wilms Tumor 1-TCR (via CRISPR) | Intellia Therapeutics | NCT05066165 [64] |
2.1.3. Role of CRISPR-Cas9 Tools in Design of Allogenic ”Off-the-Shelf” T Cell Therapies
| Conditions Targeted | Targets Knocked out via CRISPR | Targets Knocked in (via Lentivirus or CRISPR) | Sponsor | Clinical Trial ID |
|---|---|---|---|---|
| Acute myeloid leukemia | CD33 | None | Vor Biopharma | NCT05309733 [77] |
| Relapsed or refractory CD19+ leukemia and lymphoma | TRAC and β2M | CD19 CAR (via lentivirus) | Chinese PLA General Hospital | NCT03166878 [68] |
| Relapsed or refractory leukemia and lymphoma | CD19+ CD20 CAR or CD19+ CD22 CAR (via lentivirus) | NCT03398967 [69] | ||
| B cell acute lymphoblastic leukemia | TRAC and CD52 | CD19 CAR (via lentivirus) | Great Ormond Street Hospital | NCT04557436 [70] |
| Elapsed/refractory B cell non-Hodgkin lymphoma | TRAC and PD1 | CD19 CAR at TRAC loci (via CRISPR) | Caribou Biosciences, Inc. | NCT04637763 [50] |
| Relapsed or refractory T or B cell malignancies | TRAC, β2M and CD70 | CD70 CAR at TRAC loci (via CRISPR) | CRISPR Therapeutics AG | NCT04502446 [71] |
| Renal cell carcinoma | NCT04438083 [72] | |||
| B cell malignancy | TRAC and β2M | CD19 CAR at TRAC loci (via CRISPR) | NCT04035434 [73] | |
| Relapsed or refractory multiple myeloma | TRAC and β2M | BCMA CAR at TRAC loci (via CRISPR) | NCT04244656 [74] |
2.1.4. Using CRISPR-Cas9 Tools for Precise Insertion of CARs in T Cell Therapies
2.1.5. Role of CRISPR-Cas9 Tools to Mediate Effective CAR-T Therapy towards T Cell Malignancies
2.1.6. CRISPR-Cas9 Tools to Reduce Side Effects of CAR-T-Based Therapies
2.1.7. Role of CRISPR-Cas9 Tools in TCR Therapies
2.1.8. Gene Editing Efficiencies and Safety Profiles of CRISPR-Cas9-Edited T Cell Therapies
2.2. CRISPR-Cas9 Therapies for Other Non-Infectious Diseases
2.2.1. Cell Therapy for Sickle Cell Disease (SCD) or Transfusion-Dependent β-Thalassemia (TDT)
| Conditions Targeted | Gene Target | Edit Type | Therapeutic | Sponsor | Clinical Trial ID |
|---|---|---|---|---|---|
| Sickle cell disease or β-thalassemia | BCL11A | KO (NHEJ) | exa-cel | CRISPR Therapeutics and Vertex Pharmaceuticals | NCT03655678 [85] |
| NCT05477563 [86] | |||||
| NCT03745287 [87] | |||||
| NCT05356195 [88] | |||||
| NCT05329649 [89] | |||||
| NCT04208529 [90] | |||||
| ET-01 | EdiGene (GuangZhou) Inc. | NCT04925206 [95] | |||
| NCT04390971 [96] | |||||
| BRL-101 | Bioray Laboratories | NCT05577312 [97] | |||
| β-globin | HDR | nula-cel | Graphite Bio, Inc | NCT04819841 [100] | |
| CRISPR_SCD001 | UCLA, UC Berkeley | NCT04774536 [101] | |||
| iHSCs with corrected β-globin | ALLIFE Medical Science and Technology | NCT03728322 [102] | |||
| β-thalassemia | γ-globin promoter | KO (NHEJ) | BRL-101 | Bioray Laboratories | NCT04211480 [103] |
| EDIT-301 | Editas Medicine, Inc. | NCT05444894 [104] | |||
| Sickle cell disease | NCT04853576 [105] | ||||
| Type 1 diabeties | proprietary | VCTX210A | CRISPR Therapeutics and ViaCyte | NCT05210530 [106] | |
| Leber congenital amaurosis 10 | CEP290 | EDIT-101 | Editas Medicine, Inc. | NCT03872479 [107] | |
| Hereditary angioedema | KLKB1 (liver) | NTLA-2002 | Intellia Therapeutics | NCT05120830 [108] | |
| Duchenne muscular dystrophy | Dp427c | Exon skipping | CRD-TMH-001 | Cure Rare Diseases, Inc | NCT05514249 [109] |
2.2.2. Type 1 Diabetes
2.2.3. Leber Congenital Amaurosis 10 (LCA10)
2.2.4. Hereditary Angioedema
2.2.5. Duchenne Muscular Dystrophy
2.3. CRISPR-Cas9 Therapies for Viral Infections
2.3.1. Human Immunodeficiency Virus (HIV-1)
2.3.2. Human Papilloma Virus (HPV)
| Condition Targeted | Gene Target | Edit Type | Therapeutic | Sponsor | Clinical Trial ID |
|---|---|---|---|---|---|
| HIV-1 | HIV proviral DNA | Viral genome split (NHEJ) | EBT-101 | Excision Biotherapeutics | NCT05144386 [125] |
| NCT05143307 [126] | |||||
| HPV | E6/E7 genes of HPV16/18 | Viral genome split (NHEJ) | Talen: TALEN-HPV16 E6/E7 or TALEN-HPV18 E6/E7; CRISPR-Cas9: CRISPR/Cas9-HPV16 E6/E7T1 or CRISPR/Cas9-HPV18 E6/E7T2 | First Affiliated Hospital, Sun Yat-sen University | NCT03057912 [132] |
| Viral keratitis | HSV-1 genome | Viral genome split (NHEJ) | CRISPR/Cas9 mRNA | Shanghai BDgene Co., Ltd. | NCT04560790 [135] |
2.3.3. Viral Keratitis, Herpes Simplex Virus 1 (HSV-1)
3. Base Editing
3.1. Principles of Base Editing
3.2. Clinical Applications of Base Editing
3.2.1. In Vivo Delivery
3.2.2. Ex Vivo Delivery
4. Prime Editing
5. CRISPR and Gene Regulation
5.1. CRISPR Interference/CRISPR Activation (CRISPRi/CRISPRa)
5.2. CRISPR Epigenetic Editors
5.3. Applications of CRISPR Gene Regulation in Models of Human Disease
5.3.1. Retinitis Pigmentosa
5.3.2. Facioscapulohumeral Muscular Dystrophy
5.3.3. Cancer
5.3.4. Imprinting Diseases
5.3.5. HIV
5.4. Challenges and Future Perspectives
6. Emerging Applications of CRISPR-Cas in RNA Editing
6.1. RNA Editing
6.2. Cas RNA Endonucleases
6.3. Therapeutic Applications of RNA-Cleaving Cas Enzymes
7. Development of Large Animal Models of Human Diseases
CRISPR and Preclinical Development
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A Programmable Dual-RNA–Guided DNA Endonuclease in Adaptive Bacterial Immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef] [PubMed]
- Lieber, M.R.; Ma, Y.; Pannicke, U.; Schwarz, K. Mechanism and Regulation of Human Non-Homologous DNA End-Joining. Nat. Rev. Mol. Cell Biol. 2003, 4, 712–720. [Google Scholar] [CrossRef] [PubMed]
- Yeh, C.D.; Richardson, C.D.; Corn, J.E. Advances in Genome Editing through Control of DNA Repair Pathways. Nat. Cell Biol. 2019, 21, 1468–1478. [Google Scholar] [CrossRef] [PubMed]
- Xue, C.; Greene, E.C. DNA Repair Pathway Choices in CRISPR-Cas9-Mediated Genome Editing. Trends Genet. 2021, 37, 639–656. [Google Scholar] [CrossRef]
- Wright, A.V.; Nuñez, J.K.; Doudna, J.A. Biology and Applications of CRISPR Systems: Harnessing Nature’s Toolbox for Genome Engineering. Cell 2016, 164, 29–44. [Google Scholar] [CrossRef]
- Doudna, J.A.; Charpentier, E. The New Frontier of Genome Engineering with CRISPR-Cas9. Science 2014, 346, 1258096. [Google Scholar] [CrossRef]
- Fellmann, C.; Gowen, B.G.; Lin, P.-C.; Doudna, J.A.; Corn, J.E. Cornerstones of CRISPR–Cas in Drug Discovery and Therapy. Nat. Rev. Drug Discov. 2017, 16, 89–100. [Google Scholar] [CrossRef]
- Zhang, Y.; Malzahn, A.A.; Sretenovic, S.; Qi, Y. The Emerging and Uncultivated Potential of CRISPR Technology in Plant Science. Nat. Plants 2019, 5, 778–794. [Google Scholar] [CrossRef]
- Donohoue, P.D.; Barrangou, R.; May, A.P. Advances in Industrial Biotechnology Using CRISPR-Cas Systems. Trends Biotechnol. 2018, 36, 134–146. [Google Scholar] [CrossRef]
- Zhang, X.-H.; Tee, L.Y.; Wang, X.-G.; Huang, Q.-S.; Yang, S.-H. Off-Target Effects in CRISPR/Cas9-Mediated Genome Engineering. Mol. Ther. Nucleic Acids 2015, 4, e264. [Google Scholar] [CrossRef]
- Anzalone, A.V.; Koblan, L.W.; Liu, D.R. Genome Editing with CRISPR-Cas Nucleases, Base Editors, Transposases and Prime Editors. Nat. Biotechnol. 2020, 38, 824–844. [Google Scholar] [CrossRef]
- Qi, L.S.; Larson, M.H.; Gilbert, L.A.; Doudna, J.A.; Weissman, J.S.; Arkin, A.P.; Lim, W.A. Repurposing CRISPR as an RNA-Guided Platform for Sequence-Specific Control of Gene Expression. Cell 2013, 152, 1173–1183. [Google Scholar] [CrossRef] [PubMed]
- Perez-Pinera, P.; Kocak, D.D.; Vockley, C.M.; Adler, A.F.; Kabadi, A.M.; Polstein, L.R.; Thakore, P.I.; Glass, K.A.; Ousterout, D.G.; Leong, K.W.; et al. RNA-Guided Gene Activation by CRISPR-Cas9–Based Transcription Factors. Nat. Methods 2013, 10, 973–976. [Google Scholar] [CrossRef]
- Kampmann, M. CRISPRi and CRISPRa Screens in Mammalian Cells for Precision Biology and Medicine. ACS Chem. Biol. 2018, 13, 406–416. [Google Scholar] [CrossRef] [PubMed]
- Cox, D.B.T.; Gootenberg, J.S.; Abudayyeh, O.O.; Franklin, B.; Kellner, M.J.; Joung, J.; Zhang, F. RNA Editing with CRISPR-Cas13. Science 2017, 358, 1019–1027. [Google Scholar] [CrossRef]
- Özcan, A.; Krajeski, R.; Ioannidi, E.; Lee, B.; Gardner, A.; Makarova, K.S.; Koonin, E.V.; Abudayyeh, O.O.; Gootenberg, J.S. Programmable RNA Targeting with the Single-Protein CRISPR Effector Cas7-11. Nature 2021, 597, 720–725. [Google Scholar] [CrossRef] [PubMed]
- Shrock, E.; Güell, M. Chapter Six—CRISPR in Animals and Animal Models. In Progress in Molecular Biology and Translational Science; Torres-Ruiz, R., Rodriguez-Perales, S., Eds.; Academic Press: Cambridge, MA, USA, 2017; Volume 152, pp. 95–114. [Google Scholar] [CrossRef]
- Tu, Z.; Yang, W.; Yan, S.; Guo, X.; Li, X.-J. CRISPR/Cas9: A Powerful Genetic Engineering Tool for Establishing Large Animal Models of Neurodegenerative Diseases. Mol. Neurodegener. 2015, 10, 35. [Google Scholar] [CrossRef]
- Lim, K.R.Q.; Nguyen, Q.; Dzierlega, K.; Huang, Y.; Yokota, T. CRISPR-Generated Animal Models of Duchenne Muscular Dystrophy. Genes 2020, 11, 342. [Google Scholar] [CrossRef]
- Maeder, M.L.; Gersbach, C.A. Genome-Editing Technologies for Gene and Cell Therapy. Mol. Ther. 2016, 24, 430–446. [Google Scholar] [CrossRef] [PubMed]
- Dimitri, A.; Herbst, F.; Fraietta, J.A. Engineering the Next-Generation of CAR T-Cells with CRISPR-Cas9 Gene Editing. Mol. Cancer 2022, 21, 78. [Google Scholar] [CrossRef]
- US National Library of Medicine, ClinicalTrials.gov. Available online: https://ClinicalTrials.Gov/show/NCT05397184 (accessed on 15 December 2022).
- Rohaan, M.W.; Wilgenhof, S.; Haanen, J.B.A.G. Adoptive Cellular Therapies: The Current Landscape. Virchows Arch. 2019, 474, 449–461. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, S.A.; Restifo, N.P. Adoptive Cell Transfer as Personalized Immunotherapy for Human Cancer. Science 2015, 348, 62–68. [Google Scholar] [CrossRef] [PubMed]
- June, C.H.; O’Connor, R.S.; Kawalekar, O.U.; Ghassemi, S.; Milone, M.C. CAR T Cell Immunotherapy for Human Cancer. Science 2018, 359, 1361–1365. [Google Scholar] [CrossRef] [PubMed]
- US Food and Drug Administratio. Available online: https://www.fda.gov/Vaccines-Blood-Biologics/Cellular-Gene-Therapy-Products/Approved-Cellular-and-Gene-Therapy-Products (accessed on 15 December 2022).
- Shafer, P.; Kelly, L.M.; Hoyos, V. Cancer Therapy with TCR-Engineered T Cells: Current Strategies, Challenges, and Prospects. Front. Immunol. 2022, 13, 835762. [Google Scholar] [CrossRef] [PubMed]
- Hinrichs, C.S.; Rosenberg, S.A. Exploiting the Curative Potential of Adoptive T-Cell Therapy for Cancer. Immunol. Rev. 2014, 257, 56–71. [Google Scholar] [CrossRef] [PubMed]
- John, S.; Pulsipher, M.A.; Moskop, A.; Hu, Z.-H.; Phillips, C.L.; Hall, E.M.; Margossian, S.P.; Nikiforow, S.; Martin, P.L.; Oshrine, B.; et al. Real-World Outcomes for Pediatric and Young Adult Patients with Relapsed or Refractory (R/R) B-Cell Acute Lymphoblastic Leukemia (ALL) Treated with Tisagenlecleucel: Update from the Center for International Blood and Marrow Transplant Research (CIBMTR) Registry. Blood 2021, 138 (Suppl. S1), 428. [Google Scholar] [CrossRef]
- Jacobson, C.; Locke, F.L.; Ghobadi, A.; Miklos, D.B.; Lekakis, L.J.; Oluwole, O.O.; Lin, Y.; Hill, B.T.; Timmerman, J.M.; Deol, A.; et al. Long-Term (≥4 Year and ≥5 Year) Overall Survival (OS) By 12- and 24-Month Event-Free Survival (EFS): An Updated Analysis of ZUMA-1, the Pivotal Study of Axicabtagene Ciloleucel (Axi-Cel) in Patients (Pts) with Refractory Large B-Cell Lymphoma (LBCL). Blood 2021, 138 (Suppl. S1), 1764. [Google Scholar] [CrossRef]
- Majzner, R.G.; Mackall, C.L. Tumor Antigen Escape from CAR T-Cell Therapy. Cancer Discov. 2018, 8, 1219–1226. [Google Scholar] [CrossRef]
- Newick, K.; O’Brien, S.; Moon, E.; Albelda, S.M. CAR T Cell Therapy for Solid Tumors. Annu. Rev. Med. 2017, 68, 139–152. [Google Scholar] [CrossRef]
- Morotti, M.; Albukhari, A.; Alsaadi, A.; Artibani, M.; Brenton, J.D.; Curbishley, S.M.; Dong, T.; Dustin, M.L.; Hu, Z.; McGranahan, N.; et al. Promises and Challenges of Adoptive T-Cell Therapies for Solid Tumours. Br. J. Cancer 2021, 124, 1759–1776. [Google Scholar] [CrossRef]
- Gumber, D.; Wang, L.D. Improving CAR-T Immunotherapy: Overcoming the Challenges of T Cell Exhaustion. eBioMedicine 2022, 77, 103941. [Google Scholar] [CrossRef]
- Fleischer, L.C.; Spencer, H.T.; Raikar, S.S. Targeting T Cell Malignancies Using CAR-Based Immunotherapy: Challenges and Potential Solutions. J. Hematol. Oncol. 2019, 12, 141. [Google Scholar] [CrossRef]
- Sterner, R.C.; Sterner, R.M. CAR-T Cell Therapy: Current Limitations and Potential Strategies. Blood Cancer J. 2021, 11, 69. [Google Scholar] [CrossRef] [PubMed]
- Larson, R.C.; Maus, M.V. Recent Advances and Discoveries in the Mechanisms and Functions of CAR T Cells. Nat. Rev. Cancer 2021, 21, 145–161. [Google Scholar] [CrossRef] [PubMed]
- Pauken, K.E.; Wherry, E.J. Overcoming T Cell Exhaustion in Infection and Cancer. Trends Immunol. 2015, 36, 265–276. [Google Scholar] [CrossRef] [PubMed]
- Boussiotis, V.A. Molecular and Biochemical Aspects of the PD-1 Checkpoint Pathway. N. Engl. J. Med. 2016, 375, 1767–1778. [Google Scholar] [CrossRef] [PubMed]
- Chong, E.A.; Melenhorst, J.J.; Lacey, S.F.; Ambrose, D.E.; Gonzalez, V.; Levine, B.L.; June, C.H.; Schuster, S.J. PD-1 Blockade Modulates Chimeric Antigen Receptor (CAR)–Modified T Cells: Refueling the CAR. Blood 2017, 129, 1039–1041. [Google Scholar] [CrossRef]
- Choi, B.D.; Yu, X.; Castano, A.P.; Darr, H.; Henderson, D.B.; Bouffard, A.A.; Larson, R.C.; Scarfò, I.; Bailey, S.R.; Gerhard, G.M.; et al. CRISPR-Cas9 Disruption of PD-1 Enhances Activity of Universal EGFRvIII CAR T Cells in a Preclinical Model of Human Glioblastoma. J. Immunother. Cancer 2019, 7, 304. [Google Scholar] [CrossRef]
- Rupp, L.J.; Schumann, K.; Roybal, K.T.; Gate, R.E.; Ye, C.J.; Lim, W.A.; Marson, A. CRISPR/Cas9-Mediated PD-1 Disruption Enhances Anti-Tumor Efficacy of Human Chimeric Antigen Receptor T Cells. Sci. Rep. 2017, 7, 737. [Google Scholar] [CrossRef]
- US National Library of Medicine, ClinicalTrials.gov. Available online: https://ClinicalTrials.gov/show/NCT04417764 (accessed on 15 December 2022).
- US National Library of Medicine, ClinicalTrials.gov. Available online: https://ClinicalTrials.gov/show/NCT03081715 (accessed on 15 December 2022).
- US National Library of Medicine, ClinicalTrials.gov. Available online: https://ClinicalTrials.gov/show/NCT02793856 (accessed on 15 December 2022).
- US National Library of Medicine, ClinicalTrials.gov. Available online: https://ClinicalTrials.gov/show/NCT03044743 (accessed on 15 December 2022).
- US National Library of Medicine, ClinicalTrials.gov. Available online: https://ClinicalTrials.gov/show/NCT03545815 (accessed on 15 December 2022).
- US National Library of Medicine, ClinicalTrials.gov. Available online: https://ClinicalTrials.gov/show/NCT03747965 (accessed on 15 December 2022).
- US National Library of Medicine, ClinicalTrials.gov. Available online: https://ClinicalTrials.gov/show/NCT03399448 (accessed on 15 December 2022).
- US National Library of Medicine, ClinicalTrials.gov. Available online: https://Clinicaltrials.gov/show/NCT04637763 (accessed on 15 December 2022).
- Stadtmauer, E.A.; Fraietta, J.A.; Davis, M.M.; Cohen, A.D.; Weber, K.L.; Lancaster, E.; Mangan, P.A.; Kulikovskaya, I.; Gupta, M.; Chen, F.; et al. CRISPR-Engineered T Cells in Patients with Refractory Cancer. Science 2020, 367, eaba7365. [Google Scholar] [CrossRef]
- Lu, Y.; Xue, J.; Deng, T.; Zhou, X.; Yu, K.; Deng, L.; Huang, M.; Yi, X.; Liang, M.; Wang, Y.; et al. Safety and Feasibility of CRISPR-Edited T Cells in Patients with Refractory Non-Small-Cell Lung Cancer. Nat. Med. 2020, 26, 732–740. [Google Scholar] [CrossRef]
- Wang, Z.; Li, N.; Feng, K.; Chen, M.; Zhang, Y.; Liu, Y.; Yang, Q.; Nie, J.; Tang, N.; Zhang, X.; et al. Phase I Study of CAR-T Cells with PD-1 and TCR Disruption in Mesothelin-Positive Solid Tumors. Cell. Mol. Immunol. 2021, 18, 2188–2198. [Google Scholar] [CrossRef]
- Rapoport, A.P.; Stadtmauer, E.A.; Binder-Scholl, G.K.; Goloubeva, O.; Vogl, D.T.; Lacey, S.F.; Badros, A.Z.; Garfall, A.; Weiss, B.; Finklestein, J.; et al. NY-ESO-1–Specific TCR–Engineered T Cells Mediate Sustained Antigen-Specific Antitumor Effects in Myeloma. Nat. Med. 2015, 21, 914–921. [Google Scholar] [CrossRef]
- Robbins, P.F.; Kassim, S.H.; Tran, T.L.N.; Crystal, J.S.; Morgan, R.A.; Feldman, S.A.; Yang, J.C.; Dudley, M.E.; Wunderlich, J.R.; Sherry, R.M.; et al. A Pilot Trial Using Lymphocytes Genetically Engineered with an NY-ESO-1–Reactive T-Cell Receptor: Long-Term Follow-up and Correlates with Response. Clin. Cancer Res. 2015, 21, 1019–1027. [Google Scholar] [CrossRef]
- D’Angelo, S.P.; Melchiori, L.; Merchant, M.S.; Bernstein, D.; Glod, J.; Kaplan, R.; Grupp, S.; Tap, W.D.; Chagin, K.; Binder, G.K.; et al. Antitumor Activity Associated with Prolonged Persistence of Adoptively Transferred NY-ESO-1 C259T Cells in Synovial Sarcoma. Cancer Discov. 2018, 8, 944–957. [Google Scholar] [CrossRef] [PubMed]
- Adusumilli, P.S.; Zauderer, M.G.; Rivière, I.; Solomon, S.B.; Rusch, V.W.; O’Cearbhaill, R.E.; Zhu, A.; Cheema, W.; Chintala, N.K.; Halton, E.; et al. A Phase I Trial of Regional Mesothelin-Targeted CAR T-Cell Therapy in Patients with Malignant Pleural Disease, in Combination with the Anti–PD-1 Agent Pembrolizumab. Cancer Discov. 2021, 11, 2748–2763. [Google Scholar] [CrossRef] [PubMed]
- Palmer, D.C.; Webber, B.R.; Patel, Y.; Johnson, M.J.; Kariya, C.M.; Lahr, W.S.; Parkhurst, M.R.; Gartner, J.J.; Prickett, T.D.; Lowery, F.J.; et al. Internal Checkpoint Regulates T Cell Neoantigen Reactivity and Susceptibility to PD1 Blockade. Med 2022, 3, 682–704.e8. [Google Scholar] [CrossRef]
- US National Library of Medicine, ClinicalTrials.gov. Available online: https://ClinicalTrials.gov/show/NCT05566223 (accessed on 15 December 2022).
- US National Library of Medicine, ClinicalTrials.gov. Available online: https://ClinicalTrials.gov/show/NCT04426669 (accessed on 15 December 2022).
- US National Library of Medicine, ClinicalTrials.gov. Available online: https://ClinicalTrials.gov/show/NCT04767308 (accessed on 15 December 2022).
- US National Library of Medicine, ClinicalTrials.gov. Available online: https://ClinicalTrials.gov/show/NCT04037566 (accessed on 15 December 2022).
- US National Library of Medicine, ClinicalTrials.gov. Available online: https://ClinicalTrials.gov/show/NCT04976218 (accessed on 15 December 2022).
- US National Library of Medicine, ClinicalTrials.gov. Available online: https://ClinicalTrials.gov/show/NCT05066165 (accessed on 15 December 2022).
- Qasim, W. Allogeneic CAR T Cell Therapies for Leukemia. Am. J. Hematol. 2019, 94 (Suppl. S1), S50–S54. [Google Scholar] [CrossRef] [PubMed]
- Ottaviano, G.; Georgiadis, C.; Gkazi, S.A.; Syed, F.; Zhan, H.; Etuk, A.; Preece, R.; Chu, J.; Kubat, A.; Adams, S.; et al. Phase 1 Clinical Trial of CRISPR-Engineered CAR19 Universal T Cells for Treatment of Children with Refractory B Cell Leukemia. Sci. Transl. Med. 2022, 14, eabq3010. [Google Scholar] [CrossRef]
- Depil, S.; Duchateau, P.; Grupp, S.A.; Mufti, G.; Poirot, L. ‘Off-the-Shelf’ Allogeneic CAR T Cells: Development and Challenges. Nat. Rev. Drug Discov. 2020, 19, 185–199. [Google Scholar] [CrossRef]
- US National Library of Medicine, ClinicalTrials.gov. Available online: https://ClinicalTrials.gov/show/NCT03166878 (accessed on 15 December 2022).
- US National Library of Medicine, ClinicalTrials.gov. Available online: https://ClinicalTrials.gov/show/NCT03398967 (accessed on 15 December 2022).
- US National Library of Medicine, ClinicalTrials.gov. Available online: https://ClinicalTrials.gov/show/NCT04557436 (accessed on 15 December 2022).
- US National Library of Medicine, ClinicalTrials.gov. Available online: https://ClinicalTrials.gov/show/NCT04502446 (accessed on 15 December 2022).
- US National Library of Medicine, ClinicalTrials.gov. Available online: https://ClinicalTrials.gov/show/NCT04438083 (accessed on 15 December 2022).
- US National Library of Medicine, ClinicalTrials.gov. Available online: https://ClinicalTrials.gov/show/NCT04035434 (accessed on 15 December 2022).
- US National Library of Medicine, ClinicalTrials.gov. Available online: https://ClinicalTrials.gov/show/NCT04244656 (accessed on 15 December 2022).
- Eyquem, J.; Mansilla-Soto, J.; Giavridis, T.; van der Stegen, S.J.C.; Hamieh, M.; Cunanan, K.M.; Odak, A.; Gönen, M.; Sadelain, M. Targeting a CAR to the TRAC Locus with CRISPR/Cas9 Enhances Tumour Rejection. Nature 2017, 543, 113–117. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, S.; Nastoupil, L.J.; Essell, J.; Dsouza, L.; Hart, D.; Matsuda, E.; Satterfield, T.; Nesheiwat, T.; Hammad, A.; Davi, F.; et al. A First-in-Human Phase 1, Multicenter, Open-Label Study of CB-010, a Next-Generation CRISPR-Edited Allogeneic Anti-CD19 CAR-T Cell Therapy with a PD-1 Knockout, in Patients with Relapsed/Refractory B Cell Non-Hodgkin Lymphoma (ANTLER Study). Blood 2022, 140 (Suppl. S1), 9457–9458. [Google Scholar] [CrossRef]
- US National Library of Medicine, ClinicalTrials.gov. Available online: https://ClinicalTrials.gov/show/NCT05309733 (accessed on 15 December 2022).
- von Kalle, C.; Deichmann, A.; Schmidt, M. Vector Integration and Tumorigenesis. Hum. Gene Ther. 2014, 25, 475–481. [Google Scholar] [CrossRef]
- Dai, Z.; Mu, W.; Zhao, Y.; Jia, X.; Liu, J.; Wei, Q.; Tan, T.; Zhou, J. The Rational Development of CD5-Targeting Biepitopic CARs with Fully Human Heavy-Chain-Only Antigen Recognition Domains. Mol. Ther. 2021, 29, 2707–2722. [Google Scholar] [CrossRef]
- Iyer, S.P.; Sica, R.A.; Ho, P.J.; Hu, B.; Zain, J.; Prica, A.; Weng, W.-K.; Kim, Y.H.; Khodadoust, M.S.; Palomba, M.L.; et al. S262: The Cobalt-Lym Study of Ctx130: A Phase 1 Dose Escalation Study of Cd70-Targeted Allogeneic Crispr-Cas9–Engineered Car T Cells in Patients with Relapsed/Refractory (R/R) T-Cell Malignancies. HemaSphere 2022, 6, 163–164. [Google Scholar] [CrossRef]
- Si, J.; Shi, X.; Sun, S.; Zou, B.; Li, Y.; An, D.; Lin, X.; Gao, Y.; Long, F.; Pang, B.; et al. Hematopoietic Progenitor Kinase1 (HPK1) Mediates T Cell Dysfunction and Is a Druggable Target for T Cell-Based Immunotherapies. Cancer Cell 2020, 38, 551–566.e11. [Google Scholar] [CrossRef]
- Zhang, N.; Si, J.; Li, G.; Wang, Y.; Long, F.; Wang, T.; Song, Y.; Liao, X.; Gao, G.; Dimitrov, D.S. Decreasing HPK1 Expression in CD19 CAR-T Cells: A Novel Strategy to Overcome Challenges of Cell Therapy for Adult (r/r) B-ALL. J. Clin. Oncol. 2022, 40 (Suppl. 16), 7041. [Google Scholar] [CrossRef]
- Cossette, D.; Aiyer, S.; Kimball, C.; Luby, C.; Zarate, J.; Eng, J.; Doshi, V.; Cole, R.; Kolluri, N.; Jaligama, S.; et al. Clinical-Scale Production and Characterization of Ntla-5001—A Novel Approach to Manufacturing CRISPR/Cas9 Engineered T Cell Therapies. Blood 2021, 138 (Suppl. S1), 3881. [Google Scholar] [CrossRef]
- Qasim, W.; Zhan, H.; Samarasinghe, S.; Adams, S.; Amrolia, P.; Stafford, S.; Butler, K.; Rivat, C.; Wright, G.; Somana, K.; et al. Molecular Remission of Infant B-ALL after Infusion of Universal TALEN Gene-Edited CAR T Cells. Sci. Transl. Med. 2017, 9, eaaj2013. [Google Scholar] [CrossRef]
- US National Library of Medicine, ClinicalTrials.gov. Available online: https://ClinicalTrials.gov/show/NCT03655678 (accessed on 15 December 2022).
- US National Library of Medicine, ClinicalTrials.gov. Available online: https://ClinicalTrials.gov/show/NCT05477563 (accessed on 15 December 2022).
- US National Library of Medicine, ClinicalTrials.gov. Available online: https://ClinicalTrials.gov/show/NCT03745287 (accessed on 15 December 2022).
- US National Library of Medicine, ClinicalTrials.gov. Available online: https://ClinicalTrials.gov/show/NCT05356195 (accessed on 15 December 2022).
- US National Library of Medicine, ClinicalTrials.gov. Available online: https://ClinicalTrials.gov/show/NCT05329649 (accessed on 15 December 2022).
- US National Library of Medicine, ClinicalTrials.gov. Available online: https://ClinicalTrials.gov/show/NCT04208529 (accessed on 15 December 2022).
- Khosravi, M.A.; Abbasalipour, M.; Concordet, J.-P.; Berg, J.V.; Zeinali, S.; Arashkia, A.; Azadmanesh, K.; Buch, T.; Karimipoor, M. Targeted Deletion of BCL11A Gene by CRISPR-Cas9 System for Fetal Hemoglobin Reactivation: A Promising Approach for Gene Therapy of Beta Thalassemia Disease. Eur. J. Pharmacol. 2019, 854, 398–405. [Google Scholar] [CrossRef]
- Frangoul, H.; Altshuler, D.; Cappellini, M.D.; Chen, Y.-S.; Domm, J.; Eustace, B.K.; Foell, J.; de la Fuente, J.; Grupp, S.; Handgretinger, R.; et al. CRISPR-Cas9 Gene Editing for Sickle Cell Disease and β-Thalassemia. N. Engl. J. Med. 2021, 384, 252–260. [Google Scholar] [CrossRef]
- Frangoul, H.; Locatelli, F.; Bhatia, M.; Mapara, M.Y.; Molinari, L.; Sharma, A.; Lobitz, S.; de Montalembert, M.; Rondelli, D.; Steinberg, M.; et al. Efficacy and Safety of a Single Dose of Exagamglogene Autotemcel for Severe Sickle Cell Disease. Blood 2022, 140 (Suppl. S1), 29–31. [Google Scholar] [CrossRef]
- Locatelli, F.; Lang, P.; Li, A.; Corbacioglu, S.; de la Fuente, J.; Wall, D.A.; Liem, R.; Meisel, R.; Mapara, M.Y.; Shah, A.J.; et al. Efficacy and Safety of a Single Dose of Exagamglogene Autotemcel for Transfusion-Dependent β-Thalassemia. Blood 2022, 140 (Suppl. S1), 4899–4901. [Google Scholar] [CrossRef]
- US National Library of Medicine, ClinicalTrials.gov. Available online: https://ClinicalTrials.gov/show/NCT04925206 (accessed on 15 December 2022).
- US National Library of Medicine, ClinicalTrials.gov. Available online: https://ClinicalTrials.gov/show/NCT04390971 (accessed on 15 December 2022).
- US National Library of Medicine, ClinicalTrials.gov. Available online: https://ClinicalTrials.gov/show/NCT05577312 (accessed on 15 December 2022).
- Shi, J.; Fang, R.; Gao, Z.; Shi, Z.; Kuang, Z.; Zhang, Y.; Zhang, L.; Yang, H.; Zhang, Y.; Zou, D.; et al. Preliminary Safety and Efficacy Results of EDI001: An Investigator Initiated Trial on CRISPR/Cas9-Modified Autologous CD34+ Hematopoietic Stem and Progenitor Cells for Patients with Transfusion Dependent β-Thalassemia. Blood 2022, 140 (Suppl. S1), 10652–10653. [Google Scholar] [CrossRef]
- Fu, B.; Liao, J.; Chen, S.; Li, W.; Wang, Q.; Hu, J.; Yang, F.; Hsiao, S.; Jiang, Y.; Wang, L.; et al. CRISPR-Cas9-Mediated Gene Editing of the BCL11A Enhancer for Pediatric Β0/Β0 Transfusion-Dependent β-Thalassemia. Nat. Med. 2022, 28, 1573–1580. [Google Scholar] [CrossRef] [PubMed]
- US National Library of Medicine, ClinicalTrials.gov. Available online: https://ClinicalTrials.gov/show/NCT04819841 (accessed on 15 December 2022).
- US National Library of Medicine, ClinicalTrials.gov. Available online: https://ClinicalTrials.gov/show/NCT04774536 (accessed on 15 December 2022).
- US National Library of Medicine, ClinicalTrials.gov. Available online: https://ClinicalTrials.gov/show/NCT03728322 (accessed on 15 December 2022).
- US National Library of Medicine, ClinicalTrials.gov. Available online: https://ClinicalTrials.gov/show/NCT04211480 (accessed on 15 December 2022).
- US National Library of Medicine, ClinicalTrials.gov. Available online: https://ClinicalTrials.gov/show/NCT05444894 (accessed on 15 December 2022).
- US National Library of Medicine, ClinicalTrials.gov. Available online: https://ClinicalTrials.gov/show/NCT04853576 (accessed on 15 December 2022).
- US National Library of Medicine, ClinicalTrials.gov. Available online: https://ClinicalTrials.gov/show/NCT05210530 (accessed on 15 December 2022).
- US National Library of Medicine, ClinicalTrials.gov. Available online: https://ClinicalTrials.gov/show/NCT03872479 (accessed on 15 December 2022).
- US National Library of Medicine, ClinicalTrials.gov. Available online: https://ClinicalTrials.gov/show/NCT05120830 (accessed on 15 December 2022).
- US National Library of Medicine, ClinicalTrials.gov. Available online: https://ClinicalTrials.gov/show/NCT05514249 (accessed on 15 December 2022).
- De Dreuzy, E.; Heath, J.; Zuris, J.A.; Sousa, P.; Viswanathan, R.; Scott, S.; Da Silva, J.; Ta, T.; Capehart, S.; Wang, T.; et al. EDIT-301: An Experimental Autologous Cell Therapy Comprising Cas12a-RNP Modified MPB-CD34+ Cells for the Potential Treatment of SCD. Blood 2019, 134 (Suppl. S1), 4636. [Google Scholar] [CrossRef]
- Sousa, P.; Janoudi, T.; deDreuzy, E.; Shearman, M.S.; Zhang, K.; Chang, K.-H. Preclinical Development of EDIT301, an Autologous Cell Therapy Comprising AsCas12a-RNP Modified Mobilized Peripheral Blood-CD34+ Cells for the Potential Treatment of Transfusion Dependent Beta Thalassemia. Blood 2021, 138 (Suppl. S1), 1858. [Google Scholar] [CrossRef]
- Editas Medicine. Editas Medicine Announces Positive Safety and Efficacy Data from the First Two Patients Treated in the RUBY Trial of EDIT-301 for the Treatment of Severe Sickle Cell Disease. Press Release. Available online: https://ir.editasmedicine.com/news-releases/news-release-details/editas-medicine-announces-positive-safety-and-efficacy-data (accessed on 6 December 2022).
- Kanter, J.; Di Persio, J.F.; Leavey, P.; Shyr, D.C.; Thompson, A.A.; Porteus, M.H.; Intondi, A.; Lahiri, P.; Dever, D.P.; Petrusich, A.; et al. Trial In Progress: A First-in-Human Phase 1/2 Study of the Correction of a Single Nucleotide Mutation in Autologous HSCs (GPH101) to Convert HbS to HbA for Treating Severe SCD. In 2021 ASH Annual Meeting and Exposition; Elsevier: Amsterdam, The Netherlands, 2021. [Google Scholar]
- Graphite Bio. Graphite Bio Doses First Patient with Investigational Gene Editing Therapy GPH101 for Sickle Cell Disease [Press Release]. Available online: https://ir.graphitebio.com/press-releases/detail/76/graphite-bio-doses-first-patient-with-investigational-gene (accessed on 11 August 2022).
- Graphite Bio. Graphite Bio Announces Voluntary Pause of Phase 1/2 CEDAR Study of Nulabeglogene Autogedtemcel (Nula-Cel) for Sickle Cell Disease. [Press Release]. Available online: https://ir.graphitebio.com/press-releases/detail/84/graphite-bio-announces-voluntary-pause-of-phase-12-cedar (accessed on 5 January 2023).
- Ramzy, A.; Thompson, D.M.; Ward-Hartstonge, K.A.; Ivison, S.; Cook, L.; Garcia, R.V.; Loyal, J.; Kim, P.T.W.; Warnock, G.L.; Levings, M.K.; et al. Implanted Pluripotent Stem-Cell-Derived Pancreatic Endoderm Cells Secrete Glucose-Responsive C-Peptide in Patients with Type 1 Diabetes. Cell Stem Cell 2021, 28, 2047–2061.e5. [Google Scholar] [CrossRef]
- Shapiro, A.M.J.; Thompson, D.; Donner, T.W.; Bellin, M.D.; Hsueh, W.; Pettus, J.; Wilensky, J.; Daniels, M.; Wang, R.M.; Brandon, E.P.; et al. Insulin Expression and C-Peptide in Type 1 Diabetes Subjects Implanted with Stem Cell-Derived Pancreatic Endoderm Cells in an Encapsulation Device. Cell Rep. Med. 2021, 2, 100466. [Google Scholar] [CrossRef]
- CRISPR Theraputics. CRISPR Therapeutics and ViaCyte, Inc. Announce First Patient Dosed in Phase 1 Clinical Trial of Novel Gene-Edited Cell Replacement Therapy for Treatment of Type 1 Diabetes (T1D). [Press Release]. Available online: https://crisprtx.com/about-us/press-releases-and-presentations/crispr-therapeutics-and-viacyte-inc-announce-first-patient-dosed-in-phase-1-clinical-trial-of-novel-gene-edited-cell-replacement-therapy-for-treatment-of-type-1-diabetes-t1d (accessed on 2 February 2023).
- Perrault, I.; Delphin, N.; Hanein, S.; Gerber, S.; Dufier, J.-L.; Roche, O.; Defoort-Dhellemmes, S.; Dollfus, H.; Fazzi, E.; Munnich, A.; et al. Spectrum of NPHP6/CEP290 Mutations in Leber Congenital Amaurosis and Delineation of the Associated Phenotype. Hum. Mutat. 2007, 28, 416. [Google Scholar] [CrossRef]
- Maeder, M.L.; Stefanidakis, M.; Wilson, C.J.; Baral, R.; Barrera, L.A.; Bounoutas, G.S.; Bumcrot, D.; Chao, H.; Ciulla, D.M.; DaSilva, J.A.; et al. Development of a Gene-Editing Approach to Restore Vision Loss in Leber Congenital Amaurosis Type 10. Nat. Med. 2019, 25, 229–233. [Google Scholar] [CrossRef] [PubMed]
- Editas Medicine. Press Release: Editas Medicine Announces Clinical Data Demonstrating Proof of Concept of EDIT-101 from Phase 1/2 BRILLIANCE Trial. 2022. Available online: https://ir.editasmedicine.com/news-releases/news-release-details/editas-medicine-announces-clinical-data-demonstrating-proof (accessed on 15 December 2022).
- Longhurst, H. In Vivo CRISPR/Cas9 Editing of KLKB1 in Patients with Hereditary Angioedema: A First-in Human Study. In American College of Allergy, Asthma, and Immunology 2002 Annual Scientific Meeting; Elsevier: Amsterdam, The Netherlands, 2022. [Google Scholar]
- Intellia Theraputics. Press Release: Intellia Therapeutics Presents New Interim Data from First-in-Human Study of NTLA-2002 for the Treatment of Hereditary Angioedema (HAE) at the American College of Allergy, Asthma & Immunology 2022 Annual Scientific Meeting. 2022. Available online: https://ir.intelliatx.com/news-releases/news-release-details/intellia-therapeutics-presents-new-interim-data-first-human (accessed on 15 December 2022).
- Emery, A.E. Population Frequencies of Inherited Neuromuscular Diseases--a World Survey. Neuromuscul. Disord. NMD 1991, 1, 19–29. [Google Scholar] [CrossRef]
- US National Library of Medicine, ClinicalTrials.gov. Available online: https://ClinicalTrials.gov/show/NCT05144386 (accessed on 15 December 2022).
- US National Library of Medicine, ClinicalTrials.gov. Available online: https://ClinicalTrials.gov/show/NCT05143307 (accessed on 15 December 2022).
- Mancuso, P.; Chen, C.; Kaminski, R.; Gordon, J.; Liao, S.; Robinson, J.A.; Smith, M.D.; Liu, H.; Sariyer, I.K.; Sariyer, R.; et al. CRISPR Based Editing of SIV Proviral DNA in ART Treated Non-Human Primates. Nat. Commun. 2020, 11, 6065. [Google Scholar] [CrossRef]
- Excision BioTheraputics. Press Release: Excision BioTherapeutics Doses First Participant in EBT-101 Phase 1/2 Trial Evaluating EBT-101 as a Potential Cure for HIV, 2022. [Press Release]. Available online: https://www.globenewswire.com/news-release/2022/09/15/2516733/0/en/Excision-BioTherapeutics-Doses-First-Participant-in-EBT-101-Phase-1-2-Trial-Evaluating-EBT-101-as-a-Potential-Cure-for-HIV.html (accessed on 15 September 2022).
- US National Library of Medicine, ClinicalTrials.gov. Available online: https://ClinicalTrials.gov/show/NCT03164135 (accessed on 15 December 2022).
- Blanpain, C.; Libert, F.; Vassart, G.; Parmentier, M. CCR5 and HIV Infection. Recept. Channels 2002, 8, 19–31. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Wang, J.; Liu, Y.; Xie, L.; Su, B.; Mou, D.; Wang, L.; Liu, T.; Wang, X.; Zhang, B.; et al. CRISPR-Edited Stem Cells in a Patient with HIV and Acute Lymphocytic Leukemia. N. Engl. J. Med. 2019, 381, 1240–1247. [Google Scholar] [CrossRef] [PubMed]
- US National Library of Medicine, ClinicalTrials.gov. Available online: https://ClinicalTrials.gov/show/NCT03057912 (accessed on 15 December 2022).
- Tian, R.; Liu, J.; Fan, W.; Li, R.; Cui, Z.; Jin, Z.; Huang, Z.; Xie, H.; Li, L.; Huang, Z.; et al. Gene Knock-out Chain Reaction Enables High Disruption Efficiency of HPV18 E6/E7 Genes in Cervical Cancer Cells. Mol. Ther. Oncolytics 2022, 24, 171–179. [Google Scholar] [CrossRef]
- Chen, Y.; Jiang, H.; Wang, T.; He, D.; Tian, R.; Cui, Z.; Tian, X.; Gao, Q.; Ma, X.; Yang, J.; et al. In Vitro and in Vivo Growth Inhibition of Human Cervical Cancer Cells via Human Papillomavirus E6/E7 MRNAs’ Cleavage by CRISPR/Cas13a System. Antivir. Res. 2020, 178, 104794. [Google Scholar] [CrossRef]
- US National Library of Medicine, ClinicalTrials.gov. Available online: https://ClinicalTrials.gov/show/NCT04560790 (accessed on 15 December 2022).
- Yin, D.; Ling, S.; Wang, D.; Dai, Y.; Jiang, H.; Zhou, X.; Paludan, S.R.; Hong, J.; Cai, Y. Targeting Herpes Simplex Virus with CRISPR-Cas9 Cures Herpetic Stromal Keratitis in Mice. Nat. Biotechnol. 2021, 39, 567–577. [Google Scholar] [CrossRef]
- BDGene Theraputics. BD111 of BDgene Passed the FDA Orphan Drug Application. [151 Press Release]. Available online: https://www.bdgenetherapeutics.com/en/news/77.html (accessed on 24 June 2022).
- Komor, A.C.; Kim, Y.B.; Packer, M.S.; Zuris, J.A.; Liu, D.R. Programmable Editing of a Target Base in Genomic DNA without Double-Stranded DNA Cleavage. Nature 2016, 533, 420–424. [Google Scholar] [CrossRef]
- Nishida, K.; Arazoe, T.; Yachie, N.; Banno, S.; Kakimoto, M.; Tabata, M.; Mochizuki, M.; Miyabe, A.; Araki, M.; Hara, K.Y.; et al. Targeted Nucleotide Editing Using Hybrid Prokaryotic and Vertebrate Adaptive Immune Systems. Science 2016, 353, aaf8729. [Google Scholar] [CrossRef]
- Gaudelli, N.M.; Komor, A.C.; Rees, H.A.; Packer, M.S.; Badran, A.H.; Bryson, D.I.; Liu, D.R. Programmable Base Editing of A•T to G•C in Genomic DNA without DNA Cleavage. Nature 2017, 551, 464–471. [Google Scholar] [CrossRef]
- Anzalone, A.V.; Randolph, P.B.; Davis, J.R.; Sousa, A.A.; Koblan, L.W.; Levy, J.M.; Chen, P.J.; Wilson, C.; Newby, G.A.; Raguram, A.; et al. Search-and-Replace Genome Editing without Double-Strand Breaks or Donor DNA. Nature 2019, 576, 149–157. [Google Scholar] [CrossRef] [PubMed]
- US National Library of Medicine, ClinicalTrials.gov. Available online: https://ClinicalTrials.gov/show/NCT05398029 (accessed on 15 December 2022).
- Nordestgaard, B.G.; Chapman, M.J.; Humphries, S.E.; Ginsberg, H.N.; Masana, L.; Descamps, O.S.; Wiklund, O.; Hegele, R.A.; Raal, F.J.; Defesche, J.C.; et al. Familial Hypercholesterolaemia Is Underdiagnosed and Undertreated in the General Population: Guidance for Clinicians to Prevent Coronary Heart Disease: Consensus Statement of the European Atherosclerosis Society. Eur. Heart J. 2013, 34, 3478–3490. [Google Scholar] [CrossRef] [PubMed]
- Abifadel, M.; Varret, M.; Rabès, J.-P.; Allard, D.; Ouguerram, K.; Devillers, M.; Cruaud, C.; Benjannet, S.; Wickham, L.; Erlich, D.; et al. Mutations in PCSK9 Cause Autosomal Dominant Hypercholesterolemia. Nat. Genet. 2003, 34, 154–156. [Google Scholar] [CrossRef] [PubMed]
- Cohen, J.C.; Boerwinkle, E.; Mosley, T.H.; Hobbs, H.H. Sequence Variations in PCSK9, Low LDL, and Protection against Coronary Heart Disease. N. Engl. J. Med. 2006, 354, 1264–1272. [Google Scholar] [CrossRef]
- Rao, A.S.; Lindholm, D.; Rivas, M.A.; Knowles, J.W.; Montgomery, S.B.; Ingelsson, E. Large-Scale Phenome-Wide Association Study of PCSK9 Variants Demonstrates Protection Against Ischemic Stroke. Circ. Genom. Precis. Med. 2018, 11, e002162. [Google Scholar] [CrossRef] [PubMed]
- Raal, F.J.; Kallend, D.; Ray, K.K.; Turner, T.; Koenig, W.; Wright, R.S.; Wijngaard, P.L.J.; Curcio, D.; Jaros, M.J.; Leiter, L.A.; et al. Inclisiran for the Treatment of Heterozygous Familial Hypercholesterolemia. N. Engl. J. Med. 2020, 382, 1520–1530. [Google Scholar] [CrossRef]
- Musunuru, K.; Chadwick, A.C.; Mizoguchi, T.; Garcia, S.P.; DeNizio, J.E.; Reiss, C.W.; Wang, K.; Iyer, S.; Dutta, C.; Clendaniel, V.; et al. In Vivo CRISPR Base Editing of PCSK9 Durably Lowers Cholesterol in Primates. Nature 2021, 593, 429–434. [Google Scholar] [CrossRef]
- Lee, R.G.; Mazzola, A.M.; Braun, M.C.; Platt, C.; Vafai, S.B.; Kathiresan, S.; Rohde, E.; Bellinger, A.M.; Khera, A.V. Efficacy and Safety of an Investigational Single-Course CRISPR Base Editing Therapy Targeting PCSK9 in Non-Human Primate and Mouse Models. Circulation 2022, 147, 242–253. [Google Scholar] [CrossRef]
- Rothgangl, T.; Dennis, M.K.; Lin, P.J.C.; Oka, R.; Witzigmann, D.; Villiger, L.; Qi, W.; Hruzova, M.; Kissling, L.; Lenggenhager, D.; et al. In Vivo Adenine Base Editing of PCSK9 in Macaques Reduces LDL Cholesterol Levels. Nat. Biotechnol. 2021, 39, 949–957. [Google Scholar] [CrossRef]
- Chiesa, R.; Georgiadis, C.; Ottaviano, G.; Syed, F.; Braybrook, T.; Etuk, A.; Zhan, H.; Gkazi, S.A.; Preece, R.; Adams, S.; et al. Tvt CAR7: Phase 1 Clinical Trial of Base-Edited “Universal” CAR7 T Cells for Paediatric Relapsed/Refractory T-ALL. Blood. 2022, 140 (Suppl. 1), 4579–4580. [Google Scholar] [CrossRef]
- Georgiadis, C.; Rasaiyaah, J.; Gkazi, S.A.; Preece, R.; Etuk, A.; Christi, A.; Qasim, W. Base-Edited CAR T Cells for Combinational Therapy against T Cell Malignancies. Leukemia 2021, 35, 3466–3481. [Google Scholar] [CrossRef] [PubMed]
- UCL. World-First Use of Base-Edited CAR T-Cells to Treat Resistant Leukaemia; UCL Great Ormond Street Institute of Child Health. Available online: https://www.ucl.ac.uk/child-health/news/2022/dec/world-first-use-base-edited-car-t-cells-treat-resistant-leukaemia (accessed on 21 December 2022).
- Bain, B.J.; Bates, I.; Laffan, M.A. Dacie and Lewis Practical Haematology, 12th ed.; Elsevier: Amsterdam, The Netherlands, 2017. [Google Scholar]
- Harbottle, J.A. Immunotherapy to Get on Point with Base Editing. Drug Discov. Today 2021, 26, 2350–2357. [Google Scholar] [CrossRef]
- Gilbert, L.A.; Larson, M.H.; Morsut, L.; Liu, Z.; Brar, G.A.; Torres, S.E.; Stern-Ginossar, N.; Brandman, O.; Whitehead, E.H.; Doudna, J.A.; et al. CRISPR-Mediated Modular RNA-Guided Regulation of Transcription in Eukaryotes. Cell 2013, 154, 442–451. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, L.A.; Horlbeck, M.A.; Adamson, B.; Villalta, J.E.; Chen, Y.; Whitehead, E.H.; Guimaraes, C.; Panning, B.; Ploegh, H.L.; Bassik, M.C.; et al. Genome-Scale CRISPR-Mediated Control of Gene Repression and Activation. Cell 2014, 159, 647–661. [Google Scholar] [CrossRef] [PubMed]
- Margolin, J.F.; Friedman, J.R.; Meyer, W.K.; Vissing, H.; Thiesen, H.J.; Rauscher, F.J. 3rd. Krüppel-Associated Boxes Are Potent Transcriptional Repression Domains. Proc. Natl. Acad. Sci. USA 1994, 91, 4509–4513. [Google Scholar] [CrossRef]
- Groner, A.C.; Meylan, S.; Ciuffi, A.; Zangger, N.; Ambrosini, G.; Dénervaud, N.; Bucher, P.; Trono, D. KRAB-Zinc Finger Proteins and KAP1 Can Mediate Long-Range Transcriptional Repression through Heterochromatin Spreading. PLoS Genet. 2010, 6, e1000869. [Google Scholar] [CrossRef]
- Maeder, M.L.; Linder, S.J.; Cascio, V.M.; Fu, Y.; Ho, Q.H.; Joung, J.K. CRISPR RNA-Guided Activation of Endogenous Human Genes. Nat. Methods 2013, 10, 977–979. [Google Scholar] [CrossRef]
- Konermann, S.; Brigham, M.D.; Trevino, A.E.; Joung, J.; Abudayyeh, O.O.; Barcena, C.; Hsu, P.D.; Habib, N.; Gootenberg, J.S.; Nishimasu, H.; et al. Genome-Scale Transcriptional Activation by an Engineered CRISPR-Cas9 Complex. Nature 2015, 517, 583–588. [Google Scholar] [CrossRef]
- Chavez, A.; Scheiman, J.; Vora, S.; Pruitt, B.W.; Tuttle, M.; Iyer, E.P.R.; Lin, S.; Kiani, S.; Guzman, C.D.; Wiegand, D.J.; et al. Highly Efficient Cas9-Mediated Transcriptional Programming. Nat. Methods 2015, 12, 326–328. [Google Scholar] [CrossRef]
- Nakamura, M.; Gao, Y.; Dominguez, A.A.; Qi, L.S. CRISPR Technologies for Precise Epigenome Editing. Nat. Cell Biol. 2021, 23, 11–22. [Google Scholar] [CrossRef] [PubMed]
- McDonald, J.I.; Celik, H.; Rois, L.E.; Fishberger, G.; Fowler, T.; Rees, R.; Kramer, A.; Martens, A.; Edwards, J.R.; Challen, G.A. Reprogrammable CRISPR/Cas9-Based System for Inducing Site-Specific DNA Methylation. Biol. Open 2016, 5, 866–874. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.S.; Wu, H.; Ji, X.; Stelzer, Y.; Wu, X.; Czauderna, S.; Shu, J.; Dadon, D.; Young, R.A.; Jaenisch, R. Editing DNA Methylation in the Mammalian Genome. Cell 2016, 167, 233–247.e17. [Google Scholar] [CrossRef] [PubMed]
- Stepper, P.; Kungulovski, G.; Jurkowska, R.Z.; Chandra, T.; Krueger, F.; Reinhardt, R.; Reik, W.; Jeltsch, A.; Jurkowski, T.P. Efficient Targeted DNA Methylation with Chimeric DCas9-Dnmt3a-Dnmt3L Methyltransferase. Nucleic Acids Res. 2017, 45, 1703–1713. [Google Scholar] [CrossRef]
- Thakore, P.I.; D’Ippolito, A.M.; Song, L.; Safi, A.; Shivakumar, N.K.; Kabadi, A.M.; Reddy, T.E.; Crawford, G.E.; Gersbach, C.A. Highly Specific Epigenome Editing by CRISPR-Cas9 Repressors for Silencing of Distal Regulatory Elements. Nat. Methods 2015, 12, 1143–1149. [Google Scholar] [CrossRef]
- Hilton, I.B.; D’Ippolito, A.M.; Vockley, C.M.; Thakore, P.I.; Crawford, G.E.; Reddy, T.E.; Gersbach, C.A. Epigenome Editing by a CRISPR-Cas9-Based Acetyltransferase Activates Genes from Promoters and Enhancers. Nat. Biotechnol. 2015, 33, 510–517. [Google Scholar] [CrossRef]
- Amabile, A.; Migliara, A.; Capasso, P.; Biffi, M.; Cittaro, D.; Naldini, L.; Lombardo, A. Inheritable Silencing of Endogenous Genes by Hit-and-Run Targeted Epigenetic Editing. Cell 2016, 167, 219–232.e14. [Google Scholar] [CrossRef]
- Gumerson, J.D.; Alsufyani, A.; Yu, W.; Lei, J.; Sun, X.; Dong, L.; Wu, Z.; Li, T. Restoration of RPGR Expression in Vivo Using CRISPR/Cas9 Gene Editing. Gene Ther. 2022, 29, 81–93. [Google Scholar] [CrossRef]
- Moreno, A.M.; Fu, X.; Zhu, J.; Katrekar, D.; Shih, Y.-R.V.; Marlett, J.; Cabotaje, J.; Tat, J.; Naughton, J.; Lisowski, L.; et al. In Situ Gene Therapy via AAV-CRISPR-Cas9-Mediated Targeted Gene Regulation. Mol. Ther. J. Am. Soc. Gene Ther. 2018, 26, 1818–1827. [Google Scholar] [CrossRef]
- Chang, B.; Hawes, N.L.; Hurd, R.E.; Davisson, M.T.; Nusinowitz, S.; Heckenlively, J.R. Retinal Degeneration Mutants in the Mouse. Vision Res. 2002, 42, 517–525. [Google Scholar] [CrossRef]
- Yu, W.; Mookherjee, S.; Chaitankar, V.; Hiriyanna, S.; Kim, J.-W.; Brooks, M.; Ataeijannati, Y.; Sun, X.; Dong, L.; Li, T.; et al. Nrl Knockdown by AAV-Delivered CRISPR/Cas9 Prevents Retinal Degeneration in Mice. Nat. Commun. 2017, 8, 14716. [Google Scholar] [CrossRef]
- Zhu, J.; Ming, C.; Fu, X.; Duan, Y.; Hoang, D.A.; Rutgard, J.; Zhang, R.; Wang, W.; Hou, R.; Zhang, D.; et al. Gene and Mutation Independent Therapy via CRISPR-Cas9 Mediated Cellular Reprogramming in Rod Photoreceptors. Cell Res. 2017, 27, 830–833. [Google Scholar] [CrossRef] [PubMed]
- Humphries, M.M.; Rancourt, D.; Farrar, G.J.; Kenna, P.; Hazel, M.; Bush, R.A.; Sieving, P.A.; Sheils, D.M.; McNally, N.; Creighton, P.; et al. Retinopathy Induced in Mice by Targeted Disruption of the Rhodopsin Gene. Nat. Genet. 1997, 15, 216–219. [Google Scholar] [CrossRef]
- Athanasiou, D.; Aguila, M.; Bellingham, J.; Li, W.; McCulley, C.; Reeves, P.J.; Cheetham, M.E. The Molecular and Cellular Basis of Rhodopsin Retinitis Pigmentosa Reveals Potential Strategies for Therapy. Prog. Retin. Eye Res. 2018, 62, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Hartong, D.T.; Berson, E.L.; Dryja, T.P. Retinitis Pigmentosa. Lancet 2006, 368, 1795–1809. [Google Scholar] [CrossRef]
- Larhammar, D.; Nordström, K.; Larsson, T.A. Evolution of Vertebrate Rod and Cone Phototransduction Genes. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 2009, 364, 2867–2880. [Google Scholar] [CrossRef]
- Deenen, J.C.W.; Arnts, H.; van der Maarel, S.M.; Padberg, G.W.; Verschuuren, J.J.G.M.; Bakker, E.; Weinreich, S.S.; Verbeek, A.L.M.; van Engelen, B.G.M. Population-Based Incidence and Prevalence of Facioscapulohumeral Dystrophy. Neurology 2014, 83, 1056–1059. [Google Scholar] [CrossRef] [PubMed]
- van Deutekom, J.C.; Wijmenga, C.; van Tienhoven, E.A.; Gruter, A.M.; Hewitt, J.E.; Padberg, G.W.; van Ommen, G.J.; Hofker, M.H.; Frants, R.R. FSHD Associated DNA Rearrangements Are Due to Deletions of Integral Copies of a 3.2 Kb Tandemly Repeated Unit. Hum. Mol. Genet. 1993, 2, 2037–2042. [Google Scholar] [CrossRef]
- Wijmenga, C.; Hewitt, J.E.; Sandkuijl, L.A.; Clark, L.N.; Wright, T.J.; Dauwerse, H.G.; Gruter, A.M.; Hofker, M.H.; Moerer, P.; Williamson, R. Chromosome 4q DNA Rearrangements Associated with Facioscapulohumeral Muscular Dystrophy. Nat. Genet. 1992, 2, 26–30. [Google Scholar] [CrossRef] [PubMed]
- Himeda, C.L.; Jones, T.I.; Jones, P.L. CRISPR/DCas9-Mediated Transcriptional Inhibition Ameliorates the Epigenetic Dysregulation at D4Z4 and Represses DUX4-Fl in FSH Muscular Dystrophy. Mol. Ther. J. Am. Soc. Gene Ther. 2016, 24, 527–535. [Google Scholar] [CrossRef]
- Chen, C.-Y.; Chen, J.; He, L.; Stiles, B.L. PTEN: Tumor Suppressor and Metabolic Regulator. Front. Endocrinol. 2018, 9, 338. [Google Scholar] [CrossRef] [PubMed]
- Silver, D.P.; Livingston, D.M. Mechanisms of BRCA1 Tumor Suppression. Cancer Discov. 2012, 2, 679–684. [Google Scholar] [CrossRef]
- Foulkes, W.D.; Flanders, T.Y.; Pollock, P.M.; Hayward, N.K. The CDKN2A (P16) Gene and Human Cancer. Mol. Med. Camb. Mass 1997, 3, 5–20. [Google Scholar] [CrossRef]
- Hesson, L.B.; Cooper, W.N.; Latif, F. The Role of RASSF1A Methylation in Cancer. Dis. Markers 2007, 23, 73–87. [Google Scholar] [CrossRef]
- Ullah, U.; Andrabi, S.B.A.; Tripathi, S.K.; Dirasantha, O.; Kanduri, K.; Rautio, S.; Gross, C.C.; Lehtimäki, S.; Bala, K.; Tuomisto, J.; et al. Transcriptional Repressor HIC1 Contributes to Suppressive Function of Human Induced Regulatory T Cells. Cell Rep. 2018, 22, 2094–2106. [Google Scholar] [CrossRef]
- Arechavaleta-Velasco, F.; Perez-Juarez, C.E.; Gerton, G.L.; Diaz-Cueto, L. Progranulin and Its Biological Effects in Cancer. Med. Oncol. 2017, 34, 194. [Google Scholar] [CrossRef]
- Kleiber, K.; Strebhardt, K.; Martin, B.T. The Biological Relevance of FHL2 in Tumour Cells and Its Role as a Putative Cancer Target. Anticancer Res. 2007, 27, 55–61. [Google Scholar] [PubMed]
- Therrien, M.; Wong, A.M.; Kwan, E.; Rubin, G.M. Functional Analysis of CNK in RAS Signaling. Proc. Natl. Acad. Sci. USA 1999, 96, 13259–13263. [Google Scholar] [CrossRef]
- Bonneau, D.; Longy, M. Mutations of the Human PTEN Gene. Hum. Mutat. 2000, 16, 109–122. [Google Scholar] [CrossRef]
- Gros, C.; Fahy, J.; Halby, L.; Dufau, I.; Erdmann, A.; Gregoire, J.-M.; Ausseil, F.; Vispé, S.; Arimondo, P.B. DNA Methylation Inhibitors in Cancer: Recent and Future Approaches. Biochimie 2012, 94, 2280–2296. [Google Scholar] [CrossRef] [PubMed]
- Goel, A.; Arnold, C.N.; Niedzwiecki, D.; Carethers, J.M.; Dowell, J.M.; Wasserman, L.; Compton, C.; Mayer, R.J.; Bertagnolli, M.M.; Boland, C.R. Frequent Inactivation of PTEN by Promoter Hypermethylation in Microsatellite Instability-High Sporadic Colorectal Cancers. Cancer Res. 2004, 64, 3014–3021. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.-H.; Lee, H.S.; Kim, W.H. Promoter Methylation and Silencing of PTEN in Gastric Carcinoma. Lab. Investig. J. Tech. Methods Pathol. 2002, 82, 285–291. [Google Scholar] [CrossRef]
- García, J.M.; Silva, J.; Peña, C.; Garcia, V.; Rodríguez, R.; Cruz, M.A.; Cantos, B.; Provencio, M.; España, P.; Bonilla, F. Promoter Methylation of the PTEN Gene Is a Common Molecular Change in Breast Cancer. Genes. Chromosomes Cancer 2004, 41, 117–124. [Google Scholar] [CrossRef] [PubMed]
- Salvesen, H.B.; MacDonald, N.; Ryan, A.; Jacobs, I.J.; Lynch, E.D.; Akslen, L.A.; Das, S. PTEN Methylation Is Associated with Advanced Stage and Microsatellite Instability in Endometrial Carcinoma. Int. J. Cancer 2001, 91, 22–26. [Google Scholar] [CrossRef] [PubMed]
- Moses, C.; Nugent, F.; Waryah, C.B.; Garcia-Bloj, B.; Harvey, A.R.; Blancafort, P. Activating PTEN Tumor Suppressor Expression with the CRISPR/DCas9 System. Mol. Ther. Nucleic Acids 2019, 14, 287–300. [Google Scholar] [CrossRef]
- Saunderson, E.A.; Stepper, P.; Gomm, J.J.; Hoa, L.; Morgan, A.; Allen, M.D.; Jones, J.L.; Gribben, J.G.; Jurkowski, T.P.; Ficz, G. Hit-and-Run Epigenetic Editing Prevents Senescence Entry in Primary Breast Cells from Healthy Donors. Nat. Commun. 2017, 8, 1450. [Google Scholar] [CrossRef] [PubMed]
- Brenner, A.J.; Stampfer, M.R.; Aldaz, C.M. Increased P16 Expression with First Senescence Arrest in Human Mammary Epithelial Cells and Extended Growth Capacity with P16 Inactivation. Oncogene 1998, 17, 199–205. [Google Scholar] [CrossRef]
- Allach El Khattabi, L.; Backer, S.; Pinard, A.; Dieudonné, M.-N.; Tsatsaris, V.; Vaiman, D.; Dandolo, L.; Bloch-Gallego, E.; Jammes, H.; Barbaux, S. A Genome-Wide Search for New Imprinted Genes in the Human Placenta Identifies DSCAM as the First Imprinted Gene on Chromosome 21. Eur. J. Hum. Genet. EJHG 2019, 27, 49–60. [Google Scholar] [CrossRef]
- Eggermann, T.; Perez de Nanclares, G.; Maher, E.R.; Temple, I.K.; Tümer, Z.; Monk, D.; Mackay, D.J.G.; Grønskov, K.; Riccio, A.; Linglart, A.; et al. Imprinting Disorders: A Group of Congenital Disorders with Overlapping Patterns of Molecular Changes Affecting Imprinted Loci. Clin. Epigenetics 2015, 7, 123. [Google Scholar] [CrossRef]
- Barlow, D.P. Genomic Imprinting: A Mammalian Epigenetic Discovery Model. Annu. Rev. Genet. 2011, 45, 379–403. [Google Scholar] [CrossRef]
- Mabb, A.M.; Judson, M.C.; Zylka, M.J.; Philpot, B.D. Angelman Syndrome: Insights into Genomic Imprinting and Neurodevelopmental Phenotypes. Trends Neurosci. 2011, 34, 293–303. [Google Scholar] [CrossRef] [PubMed]
- Dagli, A.I.; Mathews, J.; Williams, C.A. Angelman Syndrome. In GeneReviews®; Adam, M.P., Everman, D.B., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J., Gripp, K.W., Amemiya, A., Eds.; University of Washington, Seattle: Seattle, WA, USA, 1993. [Google Scholar]
- Wolter, J.M.; Mao, H.; Fragola, G.; Simon, J.M.; Krantz, J.L.; Bazick, H.O.; Oztemiz, B.; Stein, J.L.; Zylka, M.J. Cas9 Gene Therapy for Angelman Syndrome Traps Ube3a-ATS Long Non-Coding RNA. Nature 2020, 587, 281–284. [Google Scholar] [CrossRef] [PubMed]
- Syding, L.A.; Nickl, P.; Kasparek, P.; Sedlacek, R. CRISPR/Cas9 Epigenome Editing Potential for Rare Imprinting Diseases: A Review. Cells 2020, 9, 993. [Google Scholar] [CrossRef]
- Siliciano, R.F.; Greene, W.C. HIV Latency. Cold Spring Harb. Perspect. Med. 2011, 1, a007096. [Google Scholar] [CrossRef] [PubMed]
- Da Costa, L.C.; Bomfim, L.M.; Dittz, U.V.T.; Velozo, C.d.A.; da Cunha, R.D.; Tanuri, A. Repression of HIV-1 Reactivation Mediated by CRISPR/DCas9-KRAB in Lymphoid and Myeloid Cell Models. Retrovirology 2022, 19, 12. [Google Scholar] [CrossRef]
- Wu, X.; Scott, D.A.; Kriz, A.J.; Chiu, A.C.; Hsu, P.D.; Dadon, D.B.; Cheng, A.W.; Trevino, A.E.; Konermann, S.; Chen, S.; et al. Genome-Wide Binding of the CRISPR Endonuclease Cas9 in Mammalian Cells. Nat. Biotechnol. 2014, 32, 670–676. [Google Scholar] [CrossRef]
- Kuscu, C.; Arslan, S.; Singh, R.; Thorpe, J.; Adli, M. Genome-Wide Analysis Reveals Characteristics of off-Target Sites Bound by the Cas9 Endonuclease. Nat. Biotechnol. 2014, 32, 677–683. [Google Scholar] [CrossRef]
- Xue, W.; Chen, S.; Yin, H.; Tammela, T.; Papagiannakopoulos, T.; Joshi, N.S.; Cai, W.; Yang, G.; Bronson, R.; Crowley, D.G.; et al. CRISPR-Mediated Direct Mutation of Cancer Genes in the Mouse Liver. Nature 2014, 514, 380–384. [Google Scholar] [CrossRef]
- Rahman, M.M.; Tollefsbol, T.O. Targeting Cancer Epigenetics with CRISPR-DCAS9: Principles and Prospects. Methods San Diego Calif. 2021, 187, 77–91. [Google Scholar] [CrossRef]
- Cebrian-Serrano, A.; Davies, B. CRISPR-Cas Orthologues and Variants: Optimizing the Repertoire, Specificity and Delivery of Genome Engineering Tools. Mamm. Genome Off. J. Int. Mamm. Genome Soc. 2017, 28, 247–261. [Google Scholar] [CrossRef]
- Wu, Z.; Yang, H.; Colosi, P. Effect of Genome Size on AAV Vector Packaging. Mol. Ther. J. Am. Soc. Gene Ther. 2010, 18, 80–86. [Google Scholar] [CrossRef]
- Büning, H.; Srivastava, A. Capsid Modifications for Targeting and Improving the Efficacy of AAV Vectors. Mol. Ther.-Methods Clin. Dev. 2019, 12, 248–265. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez-Guerrero, A.; Cosset, F.-L.; Verhoeyen, E. Lentiviral Vector Pseudotypes: Precious Tools to Improve Gene Modification of Hematopoietic Cells for Research and Gene Therapy. Viruses 2020, 12, 1016. [Google Scholar] [CrossRef]
- Yip, B.H. Recent Advances in CRISPR/Cas9 Delivery Strategies. Biomolecules 2020, 10, 839. [Google Scholar] [CrossRef] [PubMed]
- Lino, C.A.; Harper, J.C.; Carney, J.P.; Timlin, J.A. Delivering CRISPR: A Review of the Challenges and Approaches. Drug Deliv. 2018, 25, 1234–1257. [Google Scholar] [CrossRef]
- Bulcha, J.T.; Wang, Y.; Ma, H.; Tai, P.W.L.; Gao, G. Viral Vector Platforms within the Gene Therapy Landscape. Signal Transduct. Target. Ther. 2021, 6, 53. [Google Scholar] [CrossRef]
- Katti, A.; Diaz, B.J.; Caragine, C.M.; Sanjana, N.E.; Dow, L.E. CRISPR in Cancer Biology and Therapy. Nat. Rev. Cancer 2022, 22, 259–279. [Google Scholar] [CrossRef] [PubMed]
- Weltner, J.; Balboa, D.; Katayama, S.; Bespalov, M.; Krjutškov, K.; Jouhilahti, E.-M.; Trokovic, R.; Kere, J.; Otonkoski, T. Human Pluripotent Reprogramming with CRISPR Activators. Nat. Commun. 2018, 9, 2643. [Google Scholar] [CrossRef]
- Nuñez, J.K.; Chen, J.; Pommier, G.C.; Cogan, J.Z.; Replogle, J.M.; Adriaens, C.; Ramadoss, G.N.; Shi, Q.; Hung, K.L.; Samelson, A.J.; et al. Genome-Wide Programmable Transcriptional Memory by CRISPR-Based Epigenome Editing. Cell 2021, 184, 2503–2519.e17. [Google Scholar] [CrossRef]
- Catchpole, R.J.; Terns, M.P. New Type III CRISPR Variant and Programmable RNA Targeting Tool: Oh, Thank Heaven for Cas7-11. Mol. Cell 2021, 81, 4354–4356. [Google Scholar] [CrossRef]
- Bass, B.L.; Weintraub, H. An Unwinding Activity That Covalently Modifies Its Double-Stranded RNA Substrate. Cell 1988, 55, 1089–1098. [Google Scholar] [CrossRef] [PubMed]
- Greeve, J.; Navaratnam, N.; Scott, J. Characterization of the Apolipoprotein B MRNA Editing Enzyme: No Similarity to the Proposed Mechanism of RNA Editing in Kinetoplastid Protozoa. Nucleic Acids Res. 1991, 19, 3569–3576. [Google Scholar] [CrossRef]
- Savva, Y.A.; Rieder, L.E.; Reenan, R.A. The ADAR Protein Family. Genome Biol. 2012, 13, 252. [Google Scholar] [CrossRef] [PubMed]
- Zinshteyn, B.; Nishikura, K. Adenosine-to-Inosine RNA Editing. WIREs Syst. Biol. Med. 2009, 1, 202–209. [Google Scholar] [CrossRef] [PubMed]
- Salter, J.D.; Bennett, R.P.; Smith, H.C. The APOBEC Protein Family: United by Structure, Divergent in Function. Trends Biochem. Sci. 2016, 41, 578–594. [Google Scholar] [CrossRef]
- Woolf, T.; Chase, J.; Stinchcomb, D. Toward the Therapeutic Editing of Mutated RNA Sequences. Proc Natl Acad Sci USA 1995, 92, 8298–8302. [Google Scholar] [CrossRef]
- Khalil, A.M. The Genome Editing Revolution: Review. J. Genet. Eng. Biotechnol. 2020, 18, 68. [Google Scholar] [CrossRef] [PubMed]
- Damase, T.R.; Sukhovershin, R.; Boada, C.; Taraballi, F.; Pettigrew, R.I.; Cooke, J.P. The Limitless Future of RNA Therapeutics. Front. Bioeng. Biotechnol. 2021, 9, 628137. [Google Scholar] [CrossRef]
- Shmakov, S.; Abudayyeh, O.O.; Makarova, K.S.; Wolf, Y.I.; Gootenberg, J.S.; Semenova, E.; Minakhin, L.; Joung, J.; Konermann, S.; Severinov, K.; et al. Discovery and Functional Characterization of Diverse Class 2 CRISPR-Cas Systems. Mol. Cell 2015, 60, 385–397. [Google Scholar] [CrossRef]
- Abudayyeh, O.O.; Gootenberg, J.S.; Konermann, S.; Joung, J.; Slaymaker, I.M.; Cox, D.B.T.; Shmakov, S.; Makarova, K.S.; Semenova, E.; Minakhin, L.; et al. C2c2 Is a Single-Component Programmable RNA-Guided RNA-Targeting CRISPR Effector. Science 2016, 353, aaf5573. [Google Scholar] [CrossRef]
- Palaz, F.; Kalkan, A.K.; Can, Ö.; Demir, A.N.; Tozluyurt, A.; Özcan, A.; Ozsoz, M. CRISPR-Cas13 System as a Promising and Versatile Tool for Cancer Diagnosis, Therapy, and Research. ACS Synth. Biol. 2021, 10, 1245–1267. [Google Scholar] [CrossRef] [PubMed]
- Abudayyeh, O.O.; Gootenberg, J.S.; Franklin, B.; Koob, J.; Kellner, M.J.; Ladha, A.; Joung, J.; Kirchgatterer, P.; Cox, D.B.T.; Zhang, F. A Cytosine Deaminase for Programmable Single-Base RNA Editing. Science 2019, 365, 382–386. [Google Scholar] [CrossRef] [PubMed]
- Bot, J.F.; van der Oost, J.; Geijsen, N. The Double Life of CRISPR–Cas13. Curr. Opin. Biotechnol. 2022, 78, 102789. [Google Scholar] [CrossRef] [PubMed]
- van Beljouw, S.P.B.; Haagsma, A.C.; Rodríguez-Molina, A.; van den Berg, D.F.; Vink, J.N.A.; Brouns, S.J.J. The GRAMP CRISPR-Cas Effector Is an RNA Endonuclease Complexed with a Caspase-like Peptidase. Science 2021, 373, 1349–1353. [Google Scholar] [CrossRef]
- Kato, K.; Zhou, W.; Okazaki, S.; Isayama, Y.; Nishizawa, T.; Gootenberg, J.S.; Abudayyeh, O.O.; Nishimasu, H. Structure and Engineering of the Type III-E CRISPR-Cas7-11 Effector Complex. Cell 2022, 185, 2324–2337.e16. [Google Scholar] [CrossRef]
- Zang, H.; Zhang, Q.; Li, X. Non-Coding RNA Networks in Pulmonary Hypertension. Front. Genet. 2021, 12, 703860. [Google Scholar] [CrossRef]
- Kalpachidou, T.; Kummer, K.K.; Kress, M. Non-Coding RNAs in Neuropathic Pain. Neuronal Signal. 2020, 4, NS20190099. [Google Scholar] [CrossRef]
- Dinger, M.E.; Mercer, T.R.; Mattick, J.S. RNAs as Extracellular Signaling Molecules. J. Mol. Endocrinol. 2008, 40, 151–159. [Google Scholar] [CrossRef]
- Lin, Y.; Li, J.; Li, C.; Tu, Z.; Li, S.; Li, X.-J.; Yan, S. Application of CRISPR/Cas9 System in Establishing Large Animal Models. Front. Cell Dev. Biol. 2022, 10, 919155. [Google Scholar] [CrossRef]
- Zhao, J.; Lai, L.; Ji, W.; Zhou, Q. Genome Editing in Large Animals: Current Status and Future Prospects. Natl. Sci. Rev. 2019, 6, 402–420. [Google Scholar] [CrossRef]
- Zou, X.; Ouyang, H.; Yu, T.; Chen, X.; Pang, D.; Tang, X.; Chen, C. Preparation of a New Type 2 Diabetic Miniature Pig Model via the CRISPR/Cas9 System. Cell Death Dis. 2019, 10, 823. [Google Scholar] [CrossRef] [PubMed]
- Niu, Y.; Shen, B.; Cui, Y.; Chen, Y.; Wang, J.; Wang, L.; Kang, Y.; Zhao, X.; Si, W.; Li, W.; et al. Generation of Gene-Modified Cynomolgus Monkey via Cas9/RNA-Mediated Gene Targeting in One-Cell Embryos. Cell 2014, 156, 836–843. [Google Scholar] [CrossRef]
- Chen, Y.; Cui, Y.; Shen, B.; Niu, Y.; Zhao, X.; Wang, L.; Wang, J.; Li, W.; Zhou, Q.; Ji, W.; et al. Germline Acquisition of Cas9/RNA-Mediated Gene Modifications in Monkeys. Cell Res. 2015, 25, 262–265. [Google Scholar] [CrossRef]
- Chen, Y.; Zheng, Y.; Kang, Y.; Yang, W.; Niu, Y.; Guo, X.; Tu, Z.; Si, C.; Wang, H.; Xing, R.; et al. Functional Disruption of the Dystrophin Gene in Rhesus Monkey Using CRISPR/Cas9. Hum. Mol. Genet. 2015, 24, 3764–3774. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Li, S.; Li, X.-J. A CRISPR Monkey Model Unravels a Unique Function of PINK1 in Primate Brains. Mol. Neurodegener. 2019, 14, 17. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Liu, Y.; Tu, Z.; Xiao, C.; Yan, S.; Ma, X.; Guo, X.; Chen, X.; Yin, P.; Yang, Z.; et al. CRISPR/Cas9-Mediated PINK1 Deletion Leads to Neurodegeneration in Rhesus Monkeys. Cell Res. 2019, 29, 334–336. [Google Scholar] [CrossRef]
- Li, H.; Wu, S.; Ma, X.; Li, X.; Cheng, T.; Chen, Z.; Wu, J.; Lv, L.; Li, L.; Xu, L.; et al. Co-Editing PINK1 and DJ-1 Genes Via Adeno-Associated Virus-Delivered CRISPR/Cas9 System in Adult Monkey Brain Elicits Classical Parkinsonian Phenotype. Neurosci. Bull. 2021, 37, 1271–1288. [Google Scholar] [CrossRef]
- Sun, Z.; Ye, J.; Yuan, J. PINK1 Mediates Neuronal Survival in Monkey. Protein Cell 2022, 13, 4–5. [Google Scholar] [CrossRef]
- Yang, W.; Guo, X.; Tu, Z.; Chen, X.; Han, R.; Liu, Y.; Yan, S.; Wang, Q.; Wang, Z.; Zhao, X.; et al. PINK1 Kinase Dysfunction Triggers Neurodegeneration in the Primate Brain without Impacting Mitochondrial Homeostasis. Protein Cell 2022, 13, 26–46. [Google Scholar] [CrossRef]
- Kang, Y.; Zheng, B.; Shen, B.; Chen, Y.; Wang, L.; Wang, J.; Niu, Y.; Cui, Y.; Zhou, J.; Wang, H.; et al. CRISPR/Cas9-Mediated Dax1 Knockout in the Monkey Recapitulates Human AHC-HH. Hum. Mol. Genet. 2015, 24, 7255–7264. [Google Scholar] [CrossRef]
- Tu, Z.; Zhao, H.; Li, B.; Yan, S.; Wang, L.; Tang, Y.; Li, Z.; Bai, D.; Li, C.; Lin, Y.; et al. CRISPR/Cas9-Mediated Disruption of SHANK3 in Monkey Leads to Drug-Treatable Autism-like Symptoms. Hum. Mol. Genet. 2018, 28, 561–571. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Tu, Z.; Xu, H.; Yan, S.; Yan, H.; Zheng, Y.; Yang, W.; Zheng, J.; Li, Z.; Tian, R.; et al. Altered Neurogenesis and Disrupted Expression of Synaptic Proteins in Prefrontal Cortex of SHANK3-Deficient Non-Human Primate. Cell Res. 2017, 27, 1293–1297. [Google Scholar] [CrossRef]
- Hai, T.; Teng, F.; Guo, R.; Li, W.; Zhou, Q. One-Step Generation of Knockout Pigs by Zygote Injection of CRISPR/Cas System. Cell Res. 2014, 24, 372–375. [Google Scholar] [CrossRef] [PubMed]
- Whitworth, K.M.; Lee, K.; Benne, J.A.; Beaton, B.P.; Spate, L.D.; Murphy, S.L.; Samuel, M.S.; Mao, J.; O’Gorman, C.; Walters, E.M.; et al. Use of the CRISPR/Cas9 System to Produce Genetically Engineered Pigs from In Vitro-Derived Oocytes and Embryos1. Biol. Reprod. 2014, 91, 78. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Xin, J.; Fan, N.; Zou, Q.; Huang, J.; Ouyang, Z.; Zhao, Y.; Zhao, B.; Liu, Z.; Lai, S.; et al. Generation of CRISPR/Cas9-Mediated Gene-Targeted Pigs via Somatic Cell Nuclear Transfer. Cell. Mol. Life Sci. 2015, 72, 1175–1184. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Cao, C.; Huang, J.; Yao, J.; Hai, T.; Zheng, Q.; Wang, X.; Zhang, H.; Qin, G.; Cheng, J.; et al. One-Step Generation of Triple Gene-Targeted Pigs Using CRISPR/Cas9 System. Sci. Rep. 2016, 6, 20620. [Google Scholar] [CrossRef]
- Koppes, E.A.; Redel, B.K.; Johnson, M.A.; Skvorak, K.J.; Ghaloul-Gonzalez, L.; Yates, M.E.; Lewis, D.W.; Gollin, S.M.; Wu, Y.L.; Christ, S.E.; et al. A Porcine Model of Phenylketonuria Generated by CRISPR/Cas9 Genome Editing. JCI Insight 2020, 5, e141523. [Google Scholar] [CrossRef] [PubMed]
- Yan, S.; Tu, Z.; Liu, Z.; Fan, N.; Yang, H.; Yang, S.; Yang, W.; Zhao, Y.; Ouyang, Z.; Lai, C.; et al. A Huntingtin Knockin Pig Model Recapitulates Features of Selective Neurodegeneration in Huntington’s Disease. Cell 2018, 173, 989–1002.e13. [Google Scholar] [CrossRef]
- Isakson, S.H.; Rizzardi, A.E.; Coutts, A.W.; Carlson, D.F.; Kirstein, M.N.; Fisher, J.; Vitte, J.; Williams, K.B.; Pluhar, G.E.; Dahiya, S.; et al. Genetically Engineered Minipigs Model the Major Clinical Features of Human Neurofibromatosis Type 1. Commun. Biol. 2018, 1, 158. [Google Scholar] [CrossRef]
- Zhang, B.; Wang, C.; Zhang, Y.; Jiang, Y.; Qin, Y.; Pang, D.; Zhang, G.; Liu, H.; Xie, Z.; Yuan, H.; et al. A CRISPR-Engineered Swine Model of COL2A1 Deficiency Recapitulates Altered Early Skeletal Developmental Defects in Humans. Bone 2020, 137, 115450. [Google Scholar] [CrossRef]
- Gao, H.; Zhao, C.; Xiang, X.; Li, Y.; Zhao, Y.; Li, Z.; Pan, D.; Dai, Y.; Hara, H.; Cooper, D.K.C.; et al. Production of A1,3-Galactosyltransferase and Cytidine Monophosphate-N-Acetylneuraminic Acid Hydroxylase Gene Double-Deficient Pigs by CRISPR/Cas9 and Handmade Cloning. J. Reprod. Dev. 2017, 63, 17–26. [Google Scholar] [CrossRef]
- Chuang, C.-K.; Chen, C.-H.; Huang, C.-L.; Su, Y.-H.; Peng, S.-H.; Lin, T.-Y.; Tai, H.-C.; Yang, T.-S.; Tu, C.-F. Generation of GGTA1 Mutant Pigs by Direct Pronuclear Microinjection of CRISPR/Cas9 Plasmid Vectors. Anim. Biotechnol. 2017, 28, 174–181. [Google Scholar] [CrossRef]
- Joanna, Z.; Magdalena, H.; Agnieszka, N.-T.; Jacek, J.; Ryszard, S.; Zdzisław, S.; Barbara, G.; Daniel, L. The Production of UL16-Binding Protein 1 Targeted Pigs Using CRISPR Technology. 3 Biotech 2018, 8, 70. [Google Scholar] [CrossRef]
- Yang, L.; Güell, M.; Niu, D.; George, H.; Lesha, E.; Grishin, D.; Aach, J.; Shrock, E.; Xu, W.; Poci, J.; et al. Genome-Wide Inactivation of Porcine Endogenous Retroviruses (PERVs). Science 2015, 350, 1101–1104. [Google Scholar] [CrossRef]
- Yue, Y.; Xu, W.; Kan, Y.; Zhao, H.-Y.; Zhou, Y.; Song, X.; Wu, J.; Xiong, J.; Goswami, D.; Yang, M.; et al. Extensive Germline Genome Engineering in Pigs. Nat. Biomed. Eng. 2021, 5, 134–143. [Google Scholar] [CrossRef] [PubMed]
- Porrett, P.M.; Orandi, B.J.; Kumar, V.; Houp, J.; Anderson, D.; Cozette Killian, A.; Hauptfeld-Dolejsek, V.; Martin, D.E.; Macedon, S.; Budd, N.; et al. First Clinical-Grade Porcine Kidney Xenotransplant Using a Human Decedent Model. Am. J. Transplant. 2022, 22, 1037–1053. [Google Scholar] [CrossRef] [PubMed]
- Griffith, B.P.; Goerlich, C.E.; Singh, A.K.; Rothblatt, M.; Lau, C.L.; Shah, A.; Lorber, M.; Grazioli, A.; Saharia, K.K.; Hong, S.N.; et al. Genetically Modified Porcine-to-Human Cardiac Xenotransplantation. N. Engl. J. Med. 2022, 387, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Fan, Z.; Perisse, I.V.; Cotton, C.U.; Regouski, M.; Meng, Q.; Domb, C.; Wettere, A.J.V.; Wang, Z.; Harris, A.; White, K.L.; et al. A Sheep Model of Cystic Fibrosis Generated by CRISPR/Cas9 Disruption of the CFTR Gene. JCI Insight 2018, 3, e123529. [Google Scholar] [CrossRef]
- Williams, D.K.; Pinzón, C.; Huggins, S.; Pryor, J.H.; Falck, A.; Herman, F.; Oldeschulte, J.; Chavez, M.B.; Foster, B.L.; White, S.H.; et al. Genetic Engineering a Large Animal Model of Human Hypophosphatasia in Sheep. Sci. Rep. 2018, 8, 16945. [Google Scholar] [CrossRef]
- Menchaca, A.; dos Santos-Neto, P.C.; Souza-Neves, M.; Cuadro, F.; Mulet, A.P.; Tesson, L.; Chenouard, V.; Guiffès, A.; Heslan, J.M.; Gantier, M.; et al. Otoferlin Gene Editing in Sheep via CRISPR-Assisted SsODN-Mediated Homology Directed Repair. Sci. Rep. 2020, 10, 5995. [Google Scholar] [CrossRef]
- Amoasii, L.; Hildyard, J.C.W.; Li, H.; Sanchez-Ortiz, E.; Mireault, A.; Caballero, D.; Harron, R.; Stathopoulou, T.-R.; Massey, C.; Shelton, J.M.; et al. Gene Editing Restores Dystrophin Expression in a Canine Model of Duchenne Muscular Dystrophy. Science 2018, 362, 86–91. [Google Scholar] [CrossRef]
- Cui, Y.; Niu, Y.; Zhou, J.; Chen, Y.; Cheng, Y.; Li, S.; Ai, Z.; Chu, C.; Wang, H.; Zheng, B.; et al. Generation of a Precise Oct4-HrGFP Knockin Cynomolgus Monkey Model via CRISPR/Cas9-Assisted Homologous Recombination. Cell Res. 2018, 28, 383–386. [Google Scholar] [CrossRef]
- Wu, M.; Wei, C.; Lian, Z.; Liu, R.; Zhu, C.; Wang, H.; Cao, J.; Shen, Y.; Zhao, F.; Zhang, L.; et al. Rosa26-Targeted Sheep Gene Knock-in via CRISPR-Cas9 System. Sci. Rep. 2016, 6, 24360. [Google Scholar] [CrossRef]
- Mehravar, M.; Shirazi, A.; Nazari, M.; Banan, M. Mosaicism in CRISPR/Cas9-Mediated Genome Editing. Dev. Biol. 2019, 445, 156–162. [Google Scholar] [CrossRef] [PubMed]
- Tu, Z.; Yang, W.; Yan, S.; Yin, A.; Gao, J.; Liu, X.; Zheng, Y.; Zheng, J.; Li, Z.; Yang, S.; et al. Promoting Cas9 Degradation Reduces Mosaic Mutations in Non-Human Primate Embryos. Sci. Rep. 2017, 7, 42081. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Duan, X.; An, X.; Feng, T.; Li, P.; Li, L.; Liu, J.; Wu, P.; Pan, D.; Du, X.; et al. Efficient RNA-Guided Base Editing for Disease Modeling in Pigs. Cell Discov. 2018, 4, 64. [Google Scholar] [CrossRef]
- Wang, F.; Zhang, W.; Yang, Q.; Kang, Y.; Fan, Y.; Wei, J.; Liu, Z.; Dai, S.; Li, H.; Li, Z.; et al. Generation of a Hutchinson–Gilford Progeria Syndrome Monkey Model by Base Editing. Protein Cell 2020, 11, 809–824. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Ge, W.; Li, N.; Liu, Q.; Chen, F.; Yang, X.; Huang, X.; Ouyang, Z.; Zhang, Q.; Zhao, Y.; et al. Efficient Base Editing for Multiple Genes and Loci in Pigs Using Base Editors. Nat. Commun. 2019, 10, 2852. [Google Scholar] [CrossRef] [PubMed]
- Yuan, H.; Yu, T.; Wang, L.; Yang, L.; Zhang, Y.; Liu, H.; Li, M.; Tang, X.; Liu, Z.; Li, Z.; et al. Efficient Base Editing by RNA-Guided Cytidine Base Editors (CBEs) in Pigs. Cell. Mol. Life Sci. 2020, 77, 719–733. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bhokisham, N.; Laudermilch, E.; Traeger, L.L.; Bonilla, T.D.; Ruiz-Estevez, M.; Becker, J.R. CRISPR-Cas System: The Current and Emerging Translational Landscape. Cells 2023, 12, 1103. https://doi.org/10.3390/cells12081103
Bhokisham N, Laudermilch E, Traeger LL, Bonilla TD, Ruiz-Estevez M, Becker JR. CRISPR-Cas System: The Current and Emerging Translational Landscape. Cells. 2023; 12(8):1103. https://doi.org/10.3390/cells12081103
Chicago/Turabian StyleBhokisham, Narendranath, Ethan Laudermilch, Lindsay L. Traeger, Tonya D. Bonilla, Mercedes Ruiz-Estevez, and Jordan R. Becker. 2023. "CRISPR-Cas System: The Current and Emerging Translational Landscape" Cells 12, no. 8: 1103. https://doi.org/10.3390/cells12081103
APA StyleBhokisham, N., Laudermilch, E., Traeger, L. L., Bonilla, T. D., Ruiz-Estevez, M., & Becker, J. R. (2023). CRISPR-Cas System: The Current and Emerging Translational Landscape. Cells, 12(8), 1103. https://doi.org/10.3390/cells12081103